
Processo de Validação de Limpeza de Produtos para a Saúde de Uso Único
Categoria: Artigo, Blog, qualificação, validação
A legislação brasileira prevê requisitos normativos a serem seguidos por todas as empresas que trabalham com Produtos para a Saúde, visando garantir a segurança e eficácia dos produtos para o uso pretendido ao qual foram desenvolvidos. Para tal, as empresas fabricantes devem seguir os requisitos regulatórios da RDC 16, de 28 de março de 2013 – que aprova o Regulamento Técnico de Boas Práticas de Fabricação de Produtos Médico e Produtos para Diagnóstico de Uso In Vitro. Esta norma preconiza requisitos de qualidade, segurança e eficácia dos processos e produtos, distribuídos em 9 capítulos.
Dentre todas as particularidades que as empresas buscam para atender os requisitos normativos, regulatórios e de Boas Práticas de Fabricação – BPF, há um tema que vem se destacando constantemente, e está relacionado aos processos de limpezas aplicados aos produtos de uso único (implantáveis, odontológicos e outros), visando garantir que todos estejam livres de contaminantes.
As indústrias entendem que a responsabilidade de garantir a eficiência das etapas de limpeza é uma prática compartilhada com todos os colaboradores, visitantes e terceiros que possam adentrar nas unidades fabris. Com isso, regras de condutas severas são adotadas e expostas para garantir que, os cuidados em relação à higiene pessoal e ambiental sejam cumpridos durante toda a rotina de trabalho, e que não gerem riscos de contaminações cruzadas durantes as execuções das atividades produtivas e de apoio, a exemplo, atividades realizadas pelo controle de qualidade, manutenção e/ ou engenharia de processo.
Em função disso, existem algumas premissas que indicam que as principais aplicações dos processos de limpeza, correspondem a busca por padronizações das técnicas aplicadas, os detalhamentos e treinamentos dos métodos, as qualificações dos equipamentos que possam estar inseridos na rotina de limpeza e também as validações de performances dos processos, aplicados no decorrer da rotina de manufatura e nos monitoramentos.
Os Métodos de Qualificação e Validação de Limpeza são executados nos dispositivos / equipamentos e nos produtos finais, com o objetivo de promover evidências que demonstrem que as etapas de limpezas realizam as remoções dos contaminantes físicos, químicos e microbiológicos, que são provenientes das etapas de manufatura e que os agentes de limpeza utilizados são efetivamente removidos, a menos que seja comprovado que os contaminantes façam parte da composição química (formulação) e que estes não prejudiquem a biocompatibilidade e performance do produto.
Diante do exposto acima, esse artigo objetiva apresentar um MODELO de um método padrão de validação de limpeza que vem sendo utilizado por Indústrias de Produtos para a Saúde. As empresas geralmente dispõem de um escopo de produtos legados de uso único e de um Sistema de Gestão da Qualidade implementado, seja ele certificado ou não. A sistemática adotada pelos fabricantes para a validação de limpeza é orientada pela Norma ISO 19227:2018 – Implants for surgery – Cleanliness of orthopedic implants – General requirements.
Antes de iniciar a etapa de validação de limpeza é necessário definir algumas premissas, conforme apresentado no fluxograma da imagem 01 abaixo.
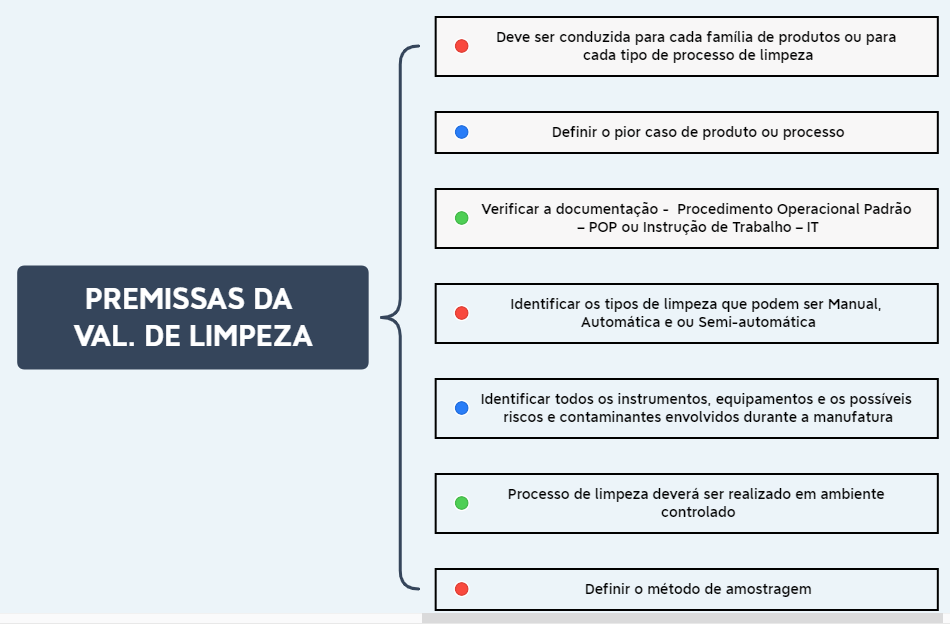
Imagem 01 – Premissas do processo de validação de limpeza
Paralelamente as premissas citadas acima, a validação de limpeza requer ainda um conhecimento amplo da equipe responsável pela atividade, no que tange o processo de manufatura dos produtos, pois é necessário identificar todos os potenciais contaminantes e potenciais interações entre os processos de produção, limpeza dos ambientes e armazenamento durante o estoque intermediário. Além disso, os métodos de limpeza empregados não devem interagir com o produto. A estratégia do ciclo de validação de limpeza para produtos legados, seguida conforme a norma ISO 19227:2018, está apresentada no fluxograma da imagem 02 a seguir:
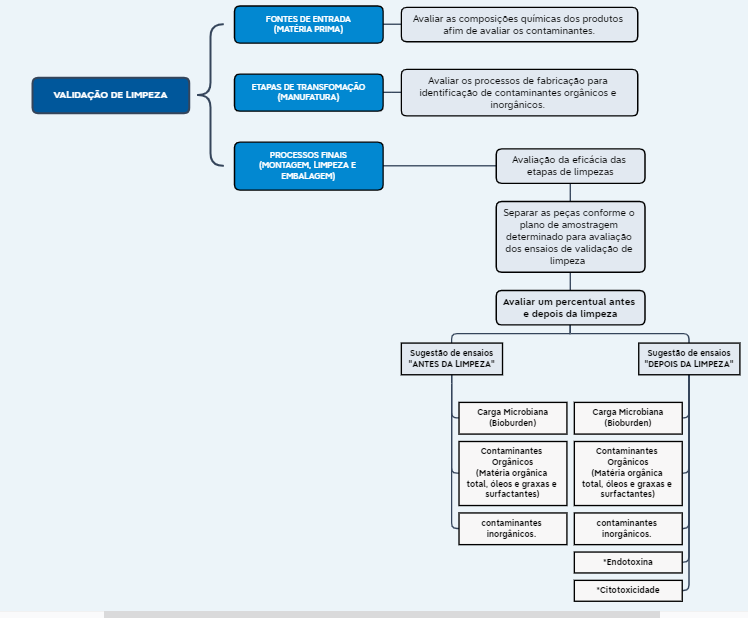
Imagem 02 – Fluxograma Conforme Sistemática da Norma ISO 19227:2018
*Os ensaios de endotoxina e citotoxicidade do produto precisam ocorrer após o processo de esterilização.
O plano de amostragem deve seguir uma abordam estatística e como sugestão podem ser utilizadas as normas ISO 2859-1:1999 – Sampling Procedures for inspection by atributes ou ABNT NBR 5426:1985 – Planos de amostragem e procedimentos na inspeção por atributos, sempre respeitando no mínimo 3 lotes consecutivos de produção contemplando a abrangência da troca de operadores e turnos de trabalho se houver necessidade, a fim de garantir a reprodutibilidade e repetibilidade dos processos.
Conforme ilustrado no fluxograma acima da imagem 02, os processos de limpeza serão avaliados em duas etapas, descritas a seguir:
Primeira etapa: Classificada como antes da limpeza do processo final, porém pode ocorrer em diversas etapas de limpeza dos produtos, a fim de garantir a efetividade de cada fase adotada para a remoção dos contaminantes. O objetivo de separar peças antes da etapa final de limpeza é avaliar a efetividade do processo geral.
Segunda etapa: Classificada como limpeza final dos produtos. É a limpeza que ocorre antes do produto ser embalado na sua embalagem final (primária e secundária). O objetivo dessa etapa é comparar as amostras da primeira etapa (sem limpeza final ou por fases) com a segunda etapa (limpeza final) e deste modo, garantir a eficiência do processo de limpeza empregado para cada produto.
Após a identificação dos tipos de etapas de separação das amostras temos que conhecer os tipos de resíduos que podem ser encontrados no decorrer dos processos. Os mesmos são classificados como:
– Resíduos solúveis orgânicos, substâncias contendo carbono;
– Resíduos solúveis inorgânicos, refere-se as substâncias contendo todos os outros elementos, exceto o carbono;
– Resíduos insolúveis/particulados, refere-se aos materiais insolúveis em meio aquoso ou solventes orgânicos que podem ser removidos sem interferir na integridade da superfície do produto.
– Resíduos Biológicos (Bactérias Mesófilas, Fungos, Leveduras e etc.).
Visando exemplificar os conceitos esclarecidos acima, a seguir será apresentado um estudo de validação de limpeza. O estudo em questão utilizou o auxílio de um equipamento automatizado, Lavadora Ultrassônica por Ultrassom, para realizar a limpeza, através do sistema de cavitação. A cavitação é a formação e colapso de milhões de bolhas minúsculas ou cavidades dentro de um líquido, produzida pela alternância de ondas de pressão altas e baixas que são geradas pelo ultrassom. As bolhas acontecem em meio ao líquido e o efeito de milhares de implosões por segundo ajudam o detergente a quebrar as moléculas de sujeira e contaminantes, resultando em uma limpeza eficaz. O processo de cavitação se dá através da utilização do equipamento “Lavadora Ultrassom”, conforme modelos apresentados abaixo no conjunto de imagens 03



Imagens 03- Modelos de Lavadoras Ultrassônicas
Por se tratar de um equipamento que tem a atividade fundamental de garantir a eficácia da limpeza, o mesmo precisa passar por todas as etapas da qualificação do equipamento, utilizando como referência a metodologia do diagrama em V (ISPE vol.5), para avaliar principalmente as qualificações de instalação, operação e desempenho do processo, pois esse estudo trata-se de um equipamento em processos de qualificação/validação concorrente ou simultânea (avaliação de rotina e monitoramento). Para equipamentos novos, a qualificação/validação será prospectiva, ou seja, antes do equipamento entrar em uso ou produto em produção e as etapas do diagrama devem ser seguidas desde o requisito do usuário, abrangendo a qualificação de projetos e assim sucessivamente, com intuito de garantir que todos os critérios estipulados pela fabricante foram atendidos e estão de acordo com as BPFs. Lembrando que todas as etapas do diagrama em V devem ser devidamente planejadas, executadas e documentadas, para evidenciar todas as etapas dos conceitos de qualificação de equipamentos. Abaixo na imagem 04 está apresentado o Diagrama em V, método universal de qualificação.
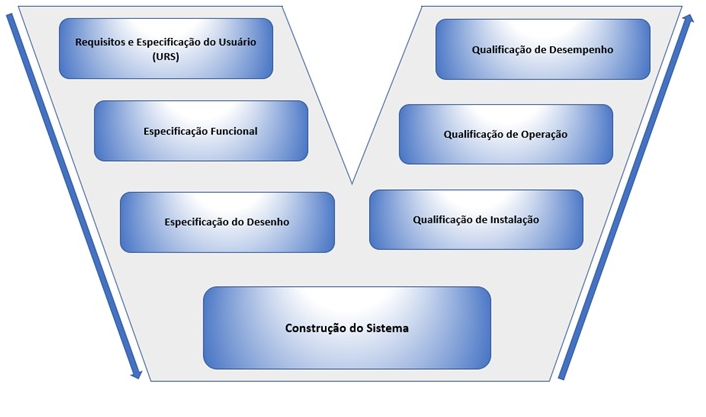
Imagem 04- Diagrama em V
A sistemática para a Qualificação do Equipamento (Lavadora Ultrassônica) apresentada abaixo será no formato de Concorrente ou Simultânea, pois trata-se de um estudo de produto legado e um equipamento que já faz parte da linha de produção.
A QUALIFICAÇÃO DE INSTALAÇÃO- QI consiste em: Avaliar se a Lavadora Ultrassônica está instalada de modo a garantir seu correto funcionamento. Nesta etapa, é necessário realizar uma checagem visual e identificar todas as informações possíveis sobre os componentes críticos do sistema, através de informações contidas nos manuais de instalação, operação e especificações técnicas do equipamento. Para comprovar se as instalações atuais estão de acordo com as especificações previstas para o equipamento, deve-se, inicialmente verificar se a voltagem do ponto de energia está adequada, avaliar as fontes de entradas das utilidades (Ex. tipo de água) e os certificados de calibração dos instrumentos de medição juntamente com as suas faixas de calibrações.
Havendo divergências, necessidades de ajustes ou correções, as mesmas devem ser tratadas como Não Conformidades, recebendo o tratamento previsto pelo sistema de gestão da qualidade da empresa, e após as devidas regularizações e finalizações do estudo de QI é necessário aprovar a etapa e evidenciar todos os registros referentes e qualificação de instalação em um relatório.
Após a Qualificação de Instalação devidamente aprovada deve ser realizada a QUALIFICAÇÃO DE OPERAÇÃO – QO. A Qualificação de Operação deverá ser realizada com base na operação do equipamento sem carga (sem produtos), buscando desafiar os limites mínimos e máximos dos parâmetros críticos. A Equipe responsável pela validação acompanhará as etapas de forma a constatar que o equipamento opera segundo os procedimentos operacionais padrões e atende todas as especificações de projeto, e também evidenciará que o pessoal envolvido encontra-se devidamente treinado. A qualificação de operação englobará também as verificações de procedimentos de manutenção preventiva e corretiva realizados por pessoal interno e ou externo, bem como os documentos de apoio aos operadores para verificar se os procedimentos estão representando a realidade dos processos e se todos os sensores e alarmes estão adequados. Do mesmo modo definido na etapa de qualificação de instalação, em caso de divergências, necessidades de ajustes ou correções, as mesmas serão tratadas como Não Conformidades, recebendo o tratamento previsto pelo sistema de gestão da qualidade da empresa. É necessário aprovar a etapa e evidenciar todos os registros referentes e qualificação de operação em um relatório que pode ser unificado com a qualificação de instalação.
Após as Qualificações de Instalação e Operação devidamente aprovadas deve ser realizada a QUALIFICAÇÃO DE DESEMPENHO OU PERFORMANCE. A Qualificação de Performance é executada com o equipamento ligado e operando com carga (produto). O objetivo é demonstrar que o processo de limpeza produz regularmente produtos dentro dos limites especificados de forma a assegurar a qualidade do produto final. O estudo inclui o acompanhamento dos ciclos de limpeza, capacidade mínima e máxima das cargas, qualidade da água do banho e a movimentação das ondas de ultra-som. Os controles críticos ocorrem através dos sensores de temperatura (ou termômetro), tempo e de velocidade que estão acoplados ou em contato com o equipamento. Na imagem 05 abaixo temos um exemplo de sistema de ultra-som de multi-estágios.
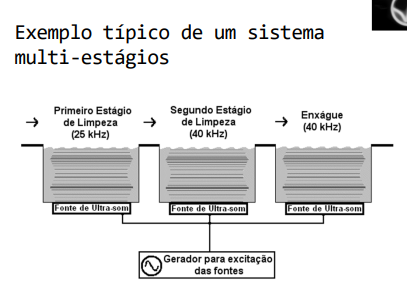
Imagem 05- Sistema da Lavadora Ultrassônica
Na imagem 06, abaixo é possível verificar a comparação entre os processos de limpeza (manual e automatizada), no qual indica que o processo de ultra-som tem um grau de limpeza e reprodutibilidade elevada em comparação com os demais processos.

Imagem 06- Comparação entre os processos de limpeza
Após a devida qualificação do equipamento é necessário elaborar a documentação com todas as evidencias de que o equipamento apresenta o desempenho consistente e reprodutível, de acordo com as especificações funcionais estabelecidas pela empresa. Uma vez qualificado o equipamento, deve-se avaliar o produto em relação aos ensaios de desempenho para validar o processo de limpeza. No fluxograma apresentado na imagem 02 acima, foram indicados ensaios de validação de desempenho do produto conforme a sistemática da ISO 19227:2018, porém os mesmos estão detalhados abaixo.
Os tipos de ensaios de performance do processo de validação de limpeza são:
– Avaliação dos Contaminantes Particulados e Inspeção visual
Os contaminantes particulados podem ser oriundos do processo de produção e/ou por contaminação cruzada. O objetivo é demonstrar que durante a limpeza os contaminantes particulados (não viáveis) foram eliminados. O método de análise utilizado deve ser por inspeção visual em lupa de aumento de 10 vezes. Esse ensaio será realizado durante o processo de inspeção visual em 100% das peças embaladas. Caso seja identificado algum desvio, será necessária a devida remoção da sujidade com o auxílio de um dispositivo (ex. Bastão de plástico e etc.). Os critérios de aceitação para a inspeção visual de contaminantes visíveis remanescentes após a limpeza devem ser seguidos conforme procedimentos operacionais padrões da empresa. Nesta etapa serão avaliadas as peças da segunda etapa (após a limpeza) e não serão retiradas amostras para ensaios.
- Ensaio de Bioburden
O ensaio de Bioburden (carga microbiana) é definido como o número de microrganismos presentes em uma superfície que não tenha sido esterilizada. Todos os produtos para a saúde exigem controle de níveis microbianos durante o processamento de limpeza e higienização. A análise de Bioburden visa medir o número total de microrganismos viáveis (Contagem total de bactérias mesófilas e contagem de fungos e leveduras) em amostra sempre antes de sua esterilização. A estimativa da população microbiana é determinada, de acordo com a norma ISO 11737-1:2018 – Sterilization of health care products Microbiological Methods – Part 1: Determination of a population of microorganisms on products. A coleta das amostras acontece em duas etapas, antes da limpeza e depois da limpeza. Para ambos é necessário utilizar a embalagem padrão (primaria e ou secundária) e os produtos devem ser identificados com sua rotulagem de rotina e também com etiqueta que identifique claramente a fase da coleta de amostra. Os resultados precisam atender as especificações dos fabricantes.
- Ensaio de Contaminantes Orgânicos
Para avaliação dos contaminantes orgânicos muitos métodos podem ser utilizados, porém serão realizados os ensaios abaixo para demonstrar a efetividade do processo:
- Matéria orgânica total (TOC) para detecção de contaminantes orgânicos solúveis em água.
- Óleos e graxas para identificar a presença de óleo e graxa (óleos lubrificantes / desmoldantes, gorduras e outros constituintes solúveis em solventes orgânicos). Os óleos e graxas são insolúveis em água, portanto a remoção total desses contaminantes em um produto pode ser crítica.
- Surfactantes (tensoativo) é uma substância utilizada para limpeza em geral, pois consegue envolver a sujeira e removê-las junto com a água, através do processo de enxague. Os surfactantes são constituídos por longas cadeias carbônicas (hidrofóbicas) com um grupo hidrofílico em uma de suas extremidades. Essa propriedade permite ao surfactante interagir tanto com substâncias polares (água) quanto com as apolares (sujeira). O excesso desses elementos no produto pode ser tóxico e causar irritações.
Com a quantificação dos contaminantes orgânicos e avaliação biológica dos dispositivos médicos, será possível estabelecer os níveis aceitáveis dos contaminantes. As peças precisam ser analisadas individualmente (antes da limpeza e depois da limpeza), sem passarem pelo processo de esterilização.
- Ensaios de Contaminantes inorgânicos
Os contaminantes inorgânicos (metais pesados) devem ser determinados após a extração exaustiva do produto, utilizando água suplementada com ácido. Quando a quantidade extraída estiver abaixo do limite de quantificação após a primeira extração, pode-se considerar que a exaustividade da extração foi demonstrada. O método de análise deve ser por varredura dos metais. Os níveis de aceitação de contaminantes inorgânicos devem ser determinados usando os dados sobre os efeitos biológicos de cada contaminante inorgânico. As peças devem ser analisadas em duas etapas (antes da limpeza e depois da limpeza), sem passarem pelo processo de esterilização e as quantidades de peças para cada ensaio são conforme definidas no plano de amostragem. As peças devem atender a avaliação dos possíveis contaminantes conforme norma estabelecida pelo fabricante.
- Ensaio de Endotoxinas Bacterianas
O teste de endotoxina bacteriana é usado para detectar ou quantificar endotoxinas de bactérias gram negativas presentes em amostras para qual o teste é preconizado. As peças precisam ser analisadas depois da limpeza e também após passarem pelo processo de esterilização. Os resultados devem atender a especificação de Endotoxina bacteriana de acordo com a norma que o fabricante determina.
- Ensaio de Citotoxicidade
A citotoxicidade dos produtos para a saúde deve ser determinada usando um método descrito na norma ISO 10993-5, Biological evaluation of medical devices — Part 5: Tests for in vitro cytotoxicit. Os produtos devem ser esterilizados. Qualquer produto que demonstre um efeito citotóxico conforme definido na norma ISO 10993-5 deve ser investigado para determinar a causa do efeito citotóxico. Se este efeito estiver relacionado à contaminação não removida pelo processo de limpeza ou ao próprio processo de limpeza, devem ser implementadas medidas de controle para mitigar esse risco. A citotoxicidade é geralmente parte da avaliação biológica, conforme a norma ISO 10993-1:2018 – Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process. É um teste in vitro útil, sensível a muitos tipos de contaminantes, que pode ser usado para avaliar a eficácia do processo de limpeza. Entretanto, mesmo se um efeito citotóxico for observado, isso pode não estar relacionado ao processo de limpeza e outras causas podem ter que ser investigadas. A etapa de coleta deve ser depois da limpeza e após passar pelo processo de esterilização e as quantidades de peças seguem conforme definidas no plano de amostragem. As peças devem atender a especificação de não ser tóxico.
Uma vez concluídas todas as etapas da qualificação do equipamento e validação do processo, a equipe responsável deverá analisar formalmente os resultados emitidos pelos laboratórios acreditados e devidamente qualificados pelo sistema de Gestão da Qualidade, e todos os resultados dos ensaios devem ser evidenciados e atender as especificações determinadas pelo fabricante ou pelas normativas utilizadas. Portanto, se atendidas às especificações em todas as etapas, os dados da avaliação de limpeza devem ser compilados em um relatório final emitido pela empresa fabricante do produto.
Todos os equipamentos e processos com status de qualificados e validados precisam ser mantidos com robustez e eficácia. Essa é uma etapa muito difícil e morosa, pois tem interface com todas as áreas e consequentemente é fundamental a parceria e colaboração de todos os envolvidos, incluindo os operadores. Para minimizar os riscos de ferir o status é necessário definir monitoramentos de rotinas. Deve-se definir em procedimento juntamente ao plano mestre de validação e qualificação (PMVQ), a periodicidade de revisão periódica do equipamento e processo que deve fornecer subsídios e evidências técnicas de que o processo é robusto e opera sem tendências negativas ou necessita de ações corretivas. As sistemáticas adotadas evidenciam a performance durante todo o ciclo de vida do equipamento e processo do produto, sempre lembrando os envolvidos da importância da responsabilidade compartilhada de atendimento das Boas Práticas de Fabricação – BPF.
Referências Bibliográficas:
– RDC N°16:2013 – Aprova o Regulamento Técnico de Boas Práticas de Fabricação de Produtos Médicos e Produtos para Diagnóstico de Uso In Vitro e dá outras providências;
– ISO 19227:2018 – Implants for surgery – Cleanliness of orthopedic implants – General requirements;
– ISO 11737-1:2018 – Sterilization of health care products Microbiological Methods – Part 1: Determination of a population of microorganisms on products;
– ISO 2859-1:1999 – Sampling Procedures for inspection by atributes;
– ABNT NBR 5426:1985 – Planos de amostragem e procedimentos na inspeção por atributos;
– ISO 10993-5, Biological evaluation of medical devices — Part 5: Tests for in vitro cytotoxicit;
– ISO 10993-1:2018 – Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process;
– Livro Qualidade 360 – O constante movimento das práticas da qualidade, da qualificação e da validação / Cordenado por Daniela Silva – Ano: 2021;
– Livro Qualificação de Equipamentos – As etapas fundamentais para garantir a qualidade do seu processo – Autora: Daniela Silva – Ano:2019.
Escrito pelos Mentorados das Mentorias Platinum e Premium – MD Consultoria (Daniela Silva)
Artigo Mentoria Platinum – Sub Grupo de Produtos para a Saúde – Maio de 2021
Antônio Junior – Químico Formado pela Universidade do Oeste Paulista (UNOESTE) e Matemático formado pela Universidade do Estado de São Paulo (UNESP). Atua na área de esterilização industrial por óxido de etileno. Experiência no campo de qualificação de equipamentos e validação de processos com ênfase em processos estéreis. Experiência na produção de produtos para saúde e sistemas de gestão da qualidade ISO 13485 e ISO 9001. Atuante da Mentoria Platinum desde 2021.
Fernanda Santos – Química Industrial formada pela PUCRS (2012). Atua na área industrial de produtos para saúde desde 2011, com experiência consolidada em sistemas de gestão da qualidade ISO 13485 e ISO 9001, gestão de produção, controle de qualidade e nos processos de Qualificações de Equipamentos e Validações de Processos Especiais e Sistemas Auxiliares. Atuante na Mentoria Platinum desde 2020.
Luciana Domingues – Farmacêutica Industrial e Bioquímica pela UBC – Universidade Braz Cubas (2010) e Licenciatura em Química com atribuições Tecnológicas pela UMC – Universidade de Mogi das Cruzes (2007). Atua na área industrial de produtos para saúde desde 2006, com experiência tratamento superficial, gestão da qualidade e processos de Qualificações e Validações. Atuante na Mentoria Platinum desde 2021.
Telma Camargo – Farmacêutica Bioquímica, graduada pela Universidade Estadual de Ponta Grossa – UEPG. Especialista em Gestão da Qualidade Six Sigma pela FAE Business School Centro Universitário. Possui mais de 20 anos de experiência em Assuntos Regulatórios, Sistema de Gestão da Qualidade, Validação de Processos Especiais e Sistemas Auxiliares, Gerenciamento de Risco, Controle e Desenvolvimento de Projetos em Dispositivos Médicos. Atuante na Mentoria Platinum desde 2019.

