
DESENVOLVIMENTO FARMACÊUTICO, CICLO DE VIDA E VALIDAÇÃO DE PROCESSOS SEGUNDO AS NOVAS EXPECTATIVAS REGULATÓRIAS NACIONAIS.
Com a publicação da RDC 301/19 a ANVISA formaliza o direcionamento regulatório na intenção de estar cada vez mais harmonizada às demais instituições sanitárias internacionais, corroborando assim com sua missão de melhorar a eficiência dos projetos de desenvolvimento e facilitar a aceitação dos produtos farmacêuticos produzidos nacionalmente em países que até então não reconheciam essa convergência regulatória. A partir de então, toda a comunidade industrial farmacêutica nacional pode evidenciar a necessidade de internalizar diretrizes internacionais, principalmente no tocante às rotinas relacionadas ao desenvolvimento de produtos e fundamentação de um efetivo, moderno e robusto sistema da qualidade.
Neste processo de interrelação é preciso entender o que de fato é desenvolvimento farmacêutico e qual estágio deste processo caracteriza o seu início, segundo entendimento da ANVISA. De uma forma simplista o processo de desenvolvimento farmacêutico se resume em: descobrir o que é crítico e controlar o que é crítico. Este processo de descoberta perpassa pelos conceitos de Gerenciamento de risco da qualidade, difundidos e pelo Q9, no qual são necessárias a Identificação do risco (o que pode dar errado), análise do risco (estimativa probabilidade x severidade), avaliação do risco (análise da conjuntura), redução do risco (redução da ocorrência ou controle do perigo) ou aceitação do risco (aceitação do risco residual em função da impossibilidade de eliminação do perigo). Em suma o estágio de desenvolvimento do processo caracteriza-se pelo risk assessment enquanto o risk control reflete o desenvolvimento das estratégias de controle. Mas a partir de qual estágio é necessário o emprego dos conceitos até então mencionados? Segundo o entendimento da Anvisa, o que caracteriza o início do desenvolvimento é o registro da Qualificação Técnica do DMF e escolha do fornecedor do Insumo Farmacêutico Ativo – IFA.
Durante o estágio de desenvolvimento farmacêutico é impossível definir o que pode ser crítico para um produto sem o entendimento de quais são os Atributos de Qualidade e os Atributos Críticos da Qualidade (CQA). Resumidamente atributos da qualidade são responsáveis por definir identidade, pureza, potência e estabilidade do produto baseado nas propriedades física, química e microbiológicas do IFA, excipientes, intermediários, embalagem primária e sistema de fechamento. Já os atributos críticos da qualidade (CQA) refletem uma propriedade ou característica física, química ou microbiológica que devem estar dentro de limites apropriados, intervalos ou distribuição para assegurar atividade, pureza e segurança desejada ao produto, como define o ICH Q8 e IN 45/19. Ilustrando o entendimento, o tamanho das partículas de um IFA pode ser um atributo da qualidade, enquanto a distribuição do tamanho destas partículas podem ser um atributo crítico da qualidade para um produto.
Entendido que no processo de desenvolvimento farmacêutico é necessário o conhecimento dos aspectos críticos relacionados ao mesmo e que este conhecimento está interrelacionado aos atributos críticos de qualidade do produto, é preciso analisar então o processo produtivo, uma vez que neste podem existir alguns parâmetros que poderão influenciar diretamente na atividade, pureza e segurança do produto. Estes são os chamados Parâmetros Críticos do Processo – CPP. Além deste, existem atributos relacionados aos materiais de entrada, que em conjunto com o CPP podem se tornar fontes consideráveis de variação dos CQA’s. Posto isto, deve-se adotar as estratégias de controles descritas na imagem a seguir com a finalidade de controlar estas fontes de variação.
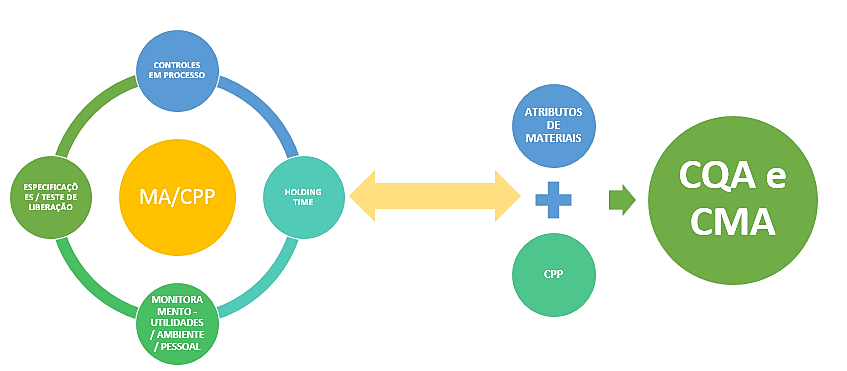
Com as novas diretrizes para as boas práticas de fabricação de medicamentos fica clara a construção dos estágios de desenvolvimento e que esta deve ser pautada no conceito de Ciclo de Vida. Neste processo os caminhos e estratégias são livres e podem perpassar tanto pela abordagem empírica, pautada no estabelecimento das amplitudes das fontes de variação através das faixas de variações conhecidas (no erro e tentativa), quanto pela abordagem de Quality By Design – QBD, baseada na utilização de ferramentas de gerenciamento de ricos ao longo da fase 1 a fim de se estabelecer as amplitudes das fontes de variação através da revisão histórica do conhecimento adquirido e experimentação, culminando na execução de estudos multivariados que se modificam a medida que o conhecimento do produto / processo é adquirido.
Com isso o mínimo que a ANVISA espera do setor regulado em inspeções de Certificação de Boas Práticas, uma vez que até então áreas de Desenvolvimento não eram inspecionadas, é a justificativa das decisões de desenvolvimento e boas práticas de documentação, para isto as empresas deverão apresentar o QTPP – Quality Target Product Profile, documento que comprova como o produto deverá ser / se comportar, em uma analogia simplificada seria a URS do produto, onde são descritos os objetivos pré-definidos para o produto e seus requerimentos de qualidade.
Neste sentido a publicação da RDC 301/19 altera radicalmente a abordagem de validação de processos produtivos de medicamentos, uma vez que todo o racional passa a ser embasado na adoção de estratégias de ciclo de vida. Neste contexto todas as fases da vida útil de um produto, equipamento e instalação, desde o desenvolvimento inicial até sua descontinuação devem ser considerados. Em seu artigo 45, a IN 47/19 resume toda expectativa da ANVISA, assim como traz nas “entrelinhas” o direcionamento necessário para as estratégias, ao descrever que a abordagem de validação de processo deve:
I -interligar o desenvolvimento de produtos e de processos; Corresponde a Fase 1 ciclo de vida
II -garantir a validação de processo de fabricação comercial; Corresponde a Fase 2 ciclo de vida
III -manter o processo em um estado de controle durante a produção comercial de rotina; Corresponde a Fase 3 ciclo de vida.
Se toda norma foi desenhada inter-relacionando conceitos definidos do ICH Q8 ao Q12, e que todos eles de uma forma indireta abordam o mesmo assunto – Risco, Gerenciamento de Risco, Conhecimento e Ciclo de Vida, os estudos de validação deverão basicamente refletir esses quatro pilares, e que para alcançar esta expectativa deverá estar correlacionado ao desenvolvimento do produto, a partir da estratégia de ciclo de vida – Fase 1, Fase 2 e Fase 3. Ilustrando temos:
Fase 1 (Desenvolvimento farmacêutico) – Desenho do produto e aquisição de conhecimento por meio de compreensão das fontes de variação. Neste estágio todo o conhecimento do produto e avaliação dos riscos nortearão a definição dos CQA’s, CMA’s e CPP’s.
Fase 2 (Qualificação do Desempenho do processo) – Confirmação do desenho do processo na escala comercial, e verificação da reprodutibilidade. Neste estágio, deverá ser desenvolvido um plano amostral que desafie a conformidade e reprodutibilidade dos CQA’s, CMA’s e CPP’s desenhados no estágio anterior, a fim de verificar e validar as estratégias de controle.
Fase 3 (Verificação continuada do processo) – Aquisição de conhecimento durante a produção comercial do produto desenvolvido de que o processo se mantém em estado de controle. Para isto, deve-se projetar o plano de amostragem desenvolvido na Fase 2, a fim de se obter segurança estatística da capabilidade do processo.
É importante considerar que o número total de lotes a ser acompanhados durante a qualificação do desempenho do processo – fase 2 está diretamente relacionada ao conhecimento prévio e informações sobre a instalação produtiva, podendo ser obtida através de avaliação de processos produtivos de produtos similares, análise de risco e informações da fase 1. Resumindo, o esforço a ser empregado no acompanhamento de lotes na fase 2 é inversamente proporcional ao conhecimento histórico adquirido através de produtos similares somado à design do produto validado, definido na fase 1.
No contexto da fase 3 o nível do esforço refletido no número de lotes acompanhados terá que ser suficiente para segurança / significância estatística, ou seja, não deve ser em pequena quantidade de modo que não se tenha robustez na análise estatística, da mesma forma que não deverá ser em grande número de modo que compromete a significância dos dados. Em suma todas estas diretrizes convergem para busca do estado de controle, e que caso em algum estágio do ciclo de vida haja um afastamento desta propriedade, ações de redesenvolvimento / melhoria contínua deverá ser imediatamente tomadas.
Para produtos legados, a verificação do estágio de controle poderá ser comprovada através dos relatórios de revisão periódica do produto considerando medidas estatísticas como a capabilidade do processo e análise dos dados da revisão periódica da qualidade. Caso o produto legado não esteja dentro de controle – os dados históricos apontam desvios, reclamações confirmadas, com origem (causa raiz) relacionada ao processo produtivo, alterações relacionadas ao processo poderão ser necessárias e neste caso, estas deverão perpassar pelo ciclo de desenvolvimento – fase 1.
Escrito por :
PEDRO HENRIQUE PIMENTA
Farmacêutico, Especialista em Regulação e Qualidade, MBA em Gestão industrial Farmacêutica,
Coordenador de Garantia da Qualidade – Qualicaps Brasil.
Membro da Mentoria Premium Daniela Silva.
REFERENCIAS
- FDA, Guidance for Industry: Process Validation – General Principles and Practices, Technical Report, 2011. https://www.fda.gov/media/71021/download
- ICH, Guideline Q8R2: Pharmaceutical Development, Technical Report, 2009. https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf
- ICH, Guideline Q9: Quality Risk Management, Technical Report, 2005. https://database.ich.org/sites/default/files/Q9%20Guideline.pdf
- ICH, Guideline Q10: Pharmaceutical Quality System, Technical Report, 2008. https://database.ich.org/sites/default/files/Q10%20Guideline.pdf
- ANVISA, Mini-Curso: Estratégia de Ciclo de Vida Aplicável à Validação de Processos (Conceitos do ICH Q8 e sua relação com a RDC 301/2019 e IN 47/2019)

